When you pick up a generic pill at the pharmacy, you expect it to work just like the brand-name version. But how does the FDA make sure it actually does? The answer lies in bioequivalence studies-rigorous tests that every generic drug maker must pass before their product hits the shelf.
Why Bioequivalence Matters
Generic drugs aren’t copies. They’re exact copies in chemical structure, but not always in how the body absorbs them. That’s where bioequivalence comes in. The FDA doesn’t just check if the active ingredient is the same. It needs proof that the generic releases the drug into your bloodstream at the same rate and to the same extent as the original. If it doesn’t, you could get too little of the drug-making it ineffective-or too much, increasing side effects.This isn’t theoretical. In 2022, nearly 90% of all prescriptions in the U.S. were filled with generics. That’s over 4 billion prescriptions a year. But if even a small fraction of those failed bioequivalence, the public health impact would be huge. That’s why the FDA treats these studies like a gatekeeper-no pass, no approval.
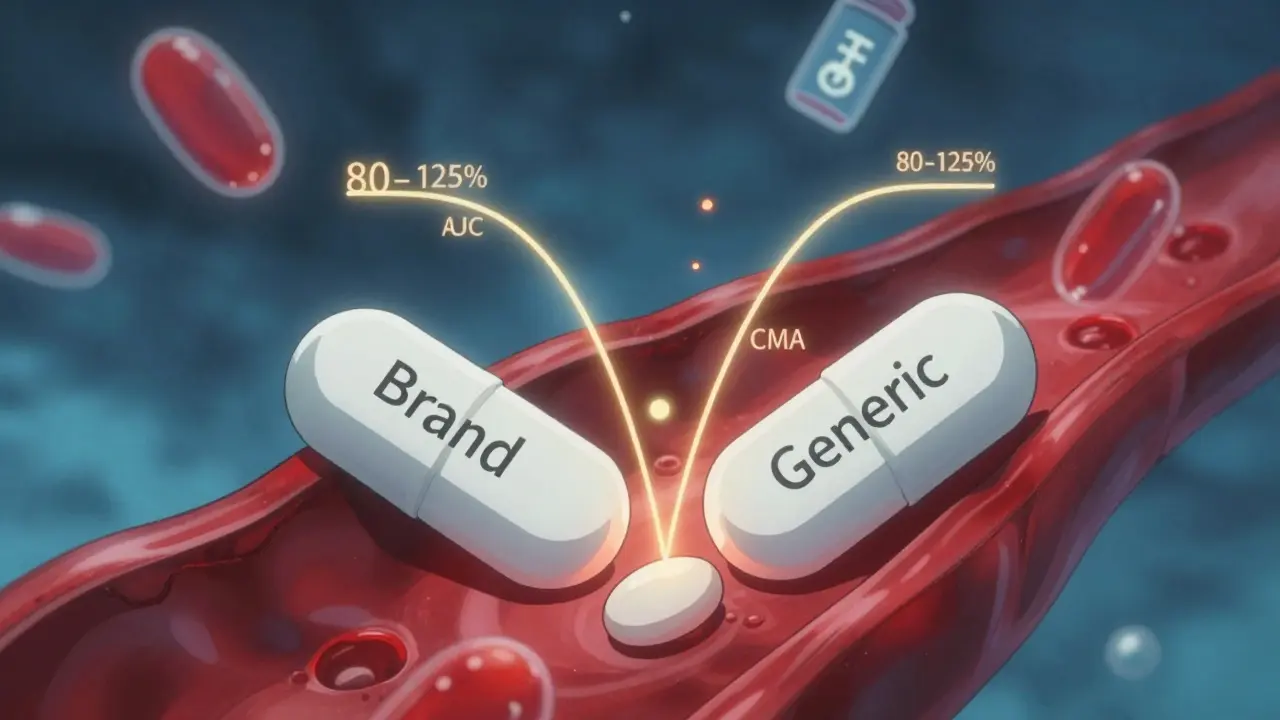
The Core Rule: 80% to 125%
The FDA’s main tool for measuring bioequivalence is a simple but powerful statistical rule: the 80-125% range. Here’s how it works.Researchers give the generic and brand-name drug to healthy volunteers-usually 24 to 36 people-and measure two key numbers: the area under the curve (AUC), which shows total drug exposure over time, and the maximum concentration (Cmax), which shows how fast the drug peaks in the blood. Both are measured in blood samples taken over several hours.
The FDA then calculates the ratio of the generic’s AUC and Cmax compared to the brand’s. If the 90% confidence interval of that ratio falls between 80% and 125%, the drugs are considered bioequivalent. That means the generic delivers between 80% and 125% of the brand’s drug levels in your body. It’s not perfect equality-but it’s close enough to guarantee the same effect.
This rule has been in place since 1992. It’s not arbitrary. It’s based on decades of clinical data showing that within this range, patients experience the same therapeutic outcomes. No more, no less.
When Fasting Isn’t Enough
Most bioequivalence studies start with volunteers fasting overnight. That’s because food can change how a drug is absorbed. But if a drug is meant to be taken with meals-like some cholesterol or diabetes meds-the FDA requires a second study under fed conditions.For example, if the brand-name drug label says “take with food,” the generic must prove it works just as well when taken after a meal. If it doesn’t, the FDA will reject it. This isn’t just paperwork. Real patients take their meds with breakfast, lunch, or dinner. The generic must behave the same way.
Some drugs are so sensitive to food that the difference between fasting and fed results can mean the difference between approval and rejection. One 2021 case involved a generic version of a blood pressure drug. The fasting study passed, but the fed study showed a 20% drop in Cmax. The manufacturer had to reformulate the tablet and resubmit-delaying approval by nine months.
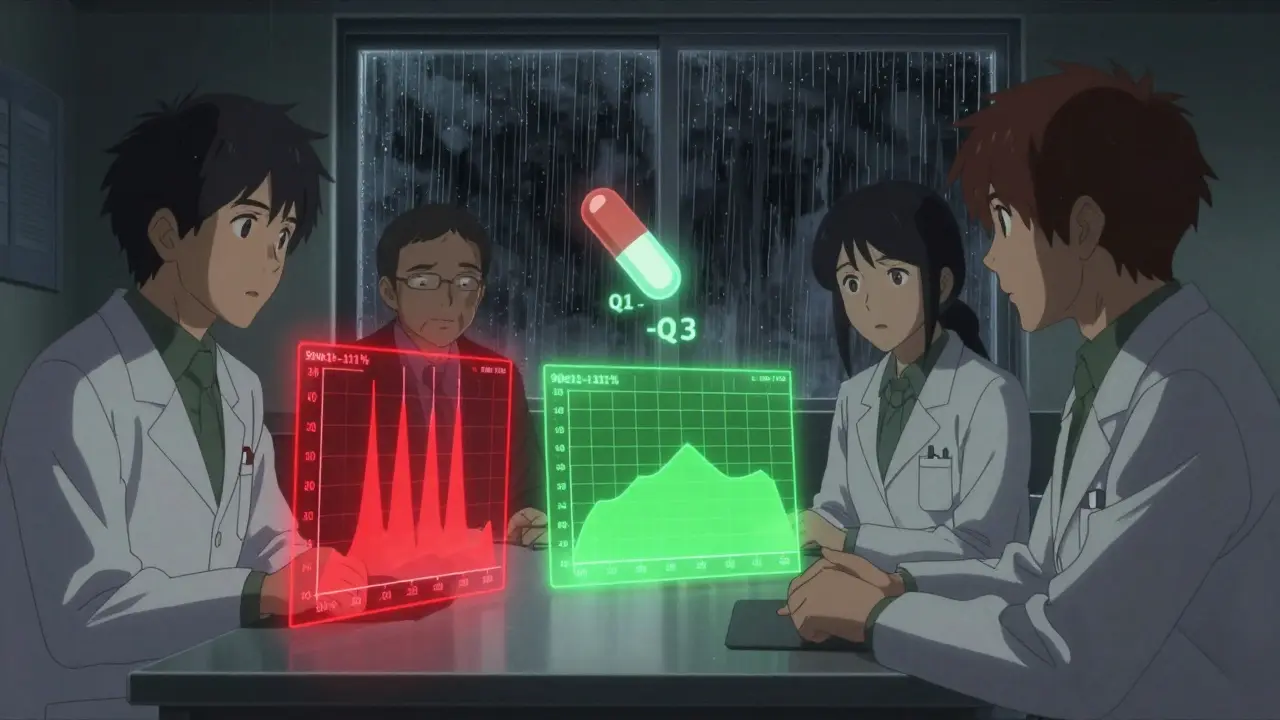
When the FDA Says: “No Human Study Needed”
Not every generic needs a full clinical trial. The FDA allows biowaivers-exceptions where bioequivalence is assumed based on science, not human testing.Biowaivers apply to a few specific cases:
- Oral solutions where the generic has the same active ingredient, concentration, and inactive ingredients as the brand.
- Topical products meant to work on the skin or eyes, not in the bloodstream-like steroid creams or eye drops.
- Inhalers with volatile anesthetics, where absorption is predictable and controlled.
To qualify, the generic must match the brand in three areas, known as the Q1-Q2-Q3 rule:
- Q1: Same active and inactive ingredients.
- Q2: Same dosage form and strength.
- Q3: Same pH, solubility, and dissolution profile.
As of late 2023, over 1,200 drug products are eligible for biowaivers. For manufacturers, this saves hundreds of thousands of dollars and 6-12 months of development time. But the FDA still requires detailed lab data proving those three criteria are met.
Tougher Rules for High-Risk Drugs
Some drugs have a narrow therapeutic index-meaning the difference between a helpful dose and a dangerous one is tiny. Warfarin, levothyroxine, and some seizure meds fall into this category.For these, the FDA tightens the bioequivalence range from 80-125% to 90-111%. That’s a much narrower window. Why? Because even a 10% difference in blood levels can cause a stroke or seizure in sensitive patients.
In 2022, the FDA rejected three generic versions of levothyroxine because their Cmax ratios fell outside the 90-111% range. Two were later approved after reformulation. One remains off the market.
For highly variable drugs-like some antibiotics or antifungals-the FDA also allows scaled average bioequivalence (SABE). This adjusts the acceptance range based on how much the brand drug’s levels vary between people. It’s a smarter, more flexible approach, but only for drugs that truly need it.
What Causes Generic Applications to Fail
Despite clear rules, nearly 60% of generic drug applications get rejected on the first try. Why?Here are the top three reasons:
- Wrong study design: Too few volunteers, poorly timed blood draws, or not testing under fed conditions when required.
- Flawed lab methods: Inaccurate drug measurements in blood samples due to poor calibration or unstable storage.
- Missing documentation: Incomplete protocols, unclear statistical analysis, or failure to follow product-specific guidances.
The FDA publishes over 2,100 product-specific guidances (PSGs) that lay out exactly how to test each drug. Companies that follow them have a 68% first-time approval rate. Those that don’t? Only 29%.
One manufacturer skipped the PSG for a generic asthma inhaler and submitted a study based on an older version of the brand. The FDA flagged 17 deficiencies. The company had to redo the entire study, costing over $1.2 million and delaying market entry by 14 months.

What’s Changing in 2026
The FDA is moving beyond traditional blood tests. New tools like physiologically based pharmacokinetic (PBPK) modeling are being used to predict how a drug behaves in the body using computer simulations. For complex products-like topical creams or inhalers-this can replace some human trials.The agency is also pushing for more U.S.-based testing. Under its Domestic Generic Drug Manufacturing Pilot Program, companies that conduct bioequivalence studies and source active ingredients in the U.S. get faster reviews. That’s a big deal: the average review time for ANDAs dropped from 18 months in 2018 to 14 months in 2023.
By mid-2024, the FDA plans to release draft guidances for 45 new complex drug categories, including transdermal patches, nasal sprays, and injectable suspensions. These will set new standards for testing, especially for drugs that don’t absorb well into the bloodstream.
What This Means for Patients
You don’t need to understand the math behind bioequivalence. But you should know this: every generic you take has been tested to make sure it works like the brand. The FDA doesn’t cut corners. It uses science, data, and strict rules to protect you.That’s why generics are safe, effective, and save Americans over $300 billion a year. The system isn’t perfect. Some studies still fail. Some companies still cut corners. But the FDA’s bioequivalence framework-backed by 40 years of experience-is the reason you can trust your $4 prescription.
What is bioequivalence and why is it important for generic drugs?
Bioequivalence means a generic drug delivers the same amount of active ingredient into your bloodstream at the same rate as the brand-name drug. It’s important because if the body absorbs the drug differently, you might not get the right effect-either too little to work, or too much and risk side effects. The FDA requires this proof before approving any generic.
Do all generic drugs need human bioequivalence studies?
No. Some generics qualify for a biowaiver, meaning no human study is needed. This applies to products like oral solutions, eye drops, or topical creams that have the same ingredients, concentration, and physical properties as the brand. The FDA uses the Q1-Q2-Q3 rule to determine eligibility. For these, lab tests and chemical analysis are enough.
What is the 80-125% rule in bioequivalence?
The 80-125% rule is the FDA’s standard for judging whether two drugs are bioequivalent. It means the 90% confidence interval of the ratio between the generic and brand drug’s AUC and Cmax must fall between 80% and 125%. If it does, the drugs are considered to have the same effect in the body. This rule has been used since 1992 and applies to most systemic drugs.
Why are bioequivalence studies for narrow therapeutic index drugs stricter?
Drugs like warfarin and levothyroxine have a narrow therapeutic index-meaning the difference between a safe dose and a toxic one is very small. A 10% difference in blood levels could cause serious harm. So the FDA tightens the acceptance range to 90-111% for these drugs to ensure even smaller variations are caught before approval.
Why do so many generic drug applications get rejected?
The most common reasons are poor study design, inaccurate lab methods, and not following the FDA’s product-specific guidances. Companies that skip these detailed instructions often face 10-20 deficiencies in their first review. Following the guidances increases first-time approval rates from 29% to 68%.
Can I trust a generic drug approved by the FDA?
Yes. Every FDA-approved generic must prove it performs the same as the brand through bioequivalence testing or a qualified biowaiver. The process is strict, data-driven, and reviewed by experts. Over 90% of prescriptions in the U.S. are generics-and they’ve been safely used for decades. The FDA’s system works because it’s built on science, not assumptions.
What Comes Next
The future of generic drugs is moving toward smarter, faster testing. Computer modeling, in vitro methods, and better analytical tools are replacing some clinical trials. But the goal stays the same: make sure every pill you take works as it should-whether it’s branded or generic.The FDA’s job isn’t to stop generics. It’s to make sure they’re safe, effective, and reliable. And for patients, that’s the only thing that matters.
 Sirolimus and Wound Healing: When to Start After Surgery
Sirolimus and Wound Healing: When to Start After Surgery
 How to Track Pediatric Doses with Apps and Dosing Charts
How to Track Pediatric Doses with Apps and Dosing Charts
 IBS-Mixed: How to Manage Alternating Constipation and Diarrhea
IBS-Mixed: How to Manage Alternating Constipation and Diarrhea
 Grifulvin V: Uses, Side Effects, and Essential Tips for Antifungal Treatment
Grifulvin V: Uses, Side Effects, and Essential Tips for Antifungal Treatment
 L-Tryptophan and Antidepressants: What You Need to Know About Serotonin Overlap and Risks
L-Tryptophan and Antidepressants: What You Need to Know About Serotonin Overlap and Risks
Jodi Harding
January 17, 2026 AT 13:15And they call this 'close enough'? Nah. Close enough is why people die quietly in their sleep.
Zoe Brooks
January 19, 2026 AT 09:08Kristin Dailey
January 20, 2026 AT 03:01Jay Clarke
January 21, 2026 AT 14:31That’s not science. That’s a magic trick with a pipette. The real question: who’s paying for these studies? Hint: not you. Not yet.
Eric Gebeke
January 23, 2026 AT 11:05And now I pay $80 a month because the system failed me. Again.
Ryan Otto
January 23, 2026 AT 19:54And now they’re pushing PBPK modeling? That’s just AI pretending to be science. You think they’re not hiding something?
Robert Cassidy
January 25, 2026 AT 04:03And the ones that slip through? They’re the ones making people sick. You think your $4 pill is safe? You’re just lucky.
Andrew Qu
January 27, 2026 AT 02:26Also - if your doctor says 'use generic,' trust them. They’ve seen the data too.
kenneth pillet
January 27, 2026 AT 09:45also why do they always use 24 people? thats like one classroom
Stacey Marsengill
January 29, 2026 AT 09:11My sister had a seizure because her generic seizure med ‘wasn’t quite right.’ The FDA said it was within 80-125%. She says it was 110% wrong.
Aysha Siera
January 31, 2026 AT 06:21rachel bellet
February 1, 2026 AT 00:30